Summary information and primary citation
- PDB-id
-
6pct;
SNAP-derived features in text and
JSON formats
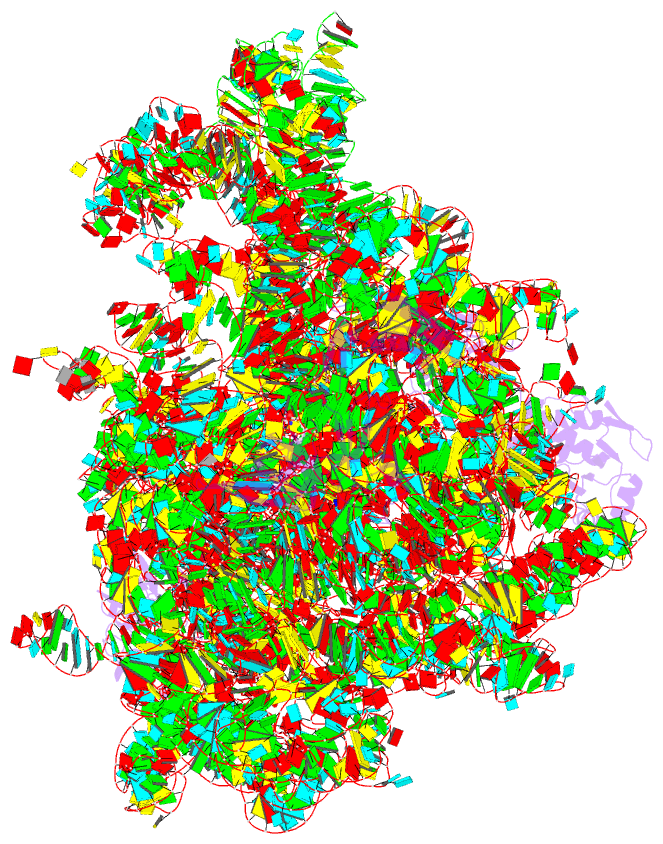
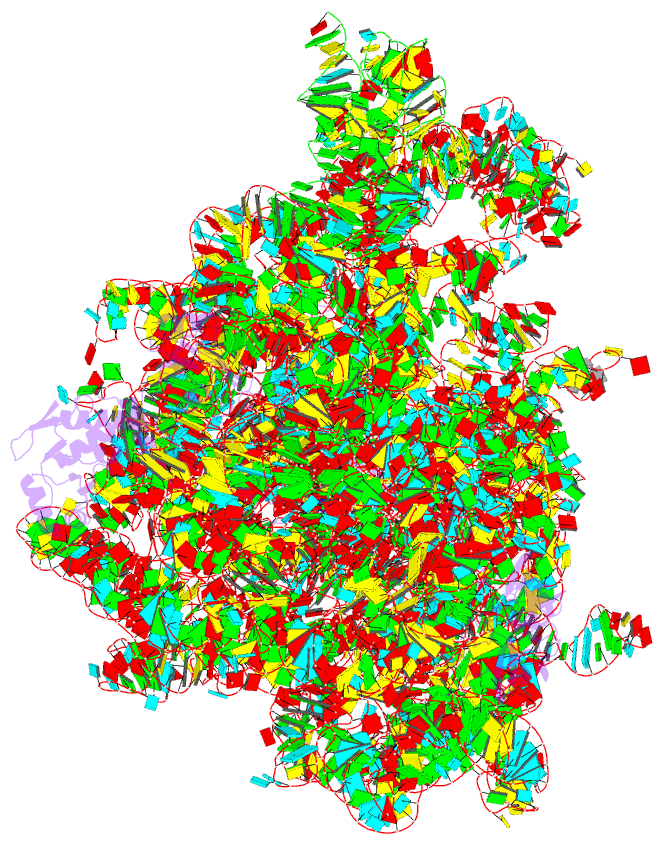
- Class
- ribosome
- Method
- cryo-EM (2.8 Å)
- Summary
- E. coli 50s ribosome bound to compound 41q
- Reference
-
Li Q, Pellegrino J, Lee DJ, Tran AA, Chaires HA, Wang R,
Park JE, Ji K, Chow D, Zhang N, Brilot AF, Biel JT, van
Zundert G, Borrelli K, Shinabarger D, Wolfe C, Murray B,
Jacobson MP, Muhle E, Chesneau O, Fraser JS, Seiple IB
(2020): "Synthetic
group A streptogramin antibiotics that overcome Vat
resistance." Nature, 586,
145-150. doi: 10.1038/s41586-020-2761-3.
- Abstract
- Natural products serve as chemical blueprints for most
antibiotics in clinical use. The evolutionary process by
which these molecules arise is inherently accompanied by
the co-evolution of resistance mechanisms that shorten the
clinical lifetime of any given class of
antibiotics<sub>1</sub>. Virginiamycin
acetyltransferase (Vat) enzymes are resistance proteins
that provide protection against
streptogramins<sub>2</sub>, potent antibiotics
against Gram-positive bacteria that inhibit the bacterial
ribosome<sub>3</sub>. Owing to the challenge of
selectively modifying the chemically complex, 23-membered
macrocyclic scaffold of group A streptogramins, analogues
that overcome the resistance conferred by Vat enzymes have
not been previously developed<sub>2</sub>. Here
we report the design, synthesis, and antibacterial
evaluation of group A streptogramin antibiotics with
extensive structural variability. Using cryo-electron
microscopy and forcefield-based refinement, we characterize
the binding of eight analogues to the bacterial ribosome at
high resolution, revealing binding interactions that extend
into the peptidyl tRNA-binding site and towards synergistic
binders that occupy the nascent peptide exit tunnel. One of
these analogues has excellent activity against several
streptogramin-resistant strains of Staphylococcus aureus,
exhibits decreased rates of acetylation in vitro, and is
effective at lowering bacterial load in a mouse model of
infection. Our results demonstrate that the combination of
rational design and modular chemical synthesis can
revitalize classes of antibiotics that are limited by
naturally arising resistance mechanisms.